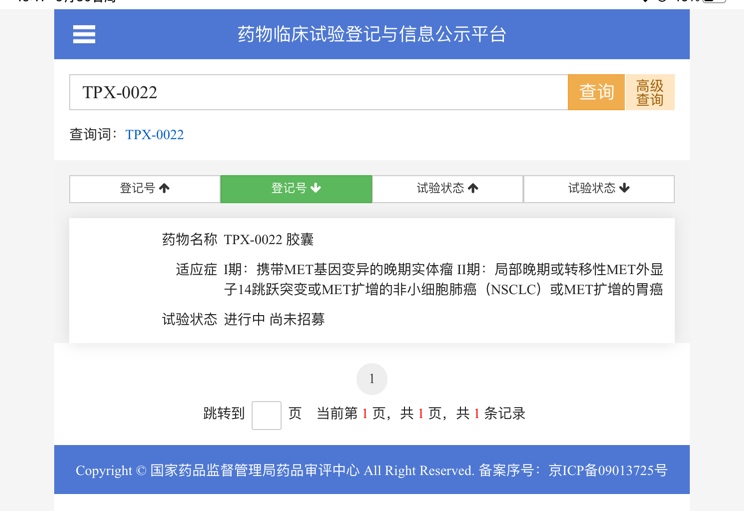
| |
 肺癌术后2年重获新生,不焦虑不内耗
讲述者:黄宇星整理者:pear编者按“用实力坚持到胜利”ALK阳性非小细胞肺癌患者摄影
肺癌术后2年重获新生,不焦虑不内耗
讲述者:黄宇星整理者:pear编者按“用实力坚持到胜利”ALK阳性非小细胞肺癌患者摄影
 基因检测结果刚出来,麻烦懂得病友帮
我母亲刚确诊是肺腺癌晚期,基因检测结果刚出来,麻烦懂的病友帮忙给看一下
基因检测结果刚出来,麻烦懂得病友帮
我母亲刚确诊是肺腺癌晚期,基因检测结果刚出来,麻烦懂的病友帮忙给看一下
 肺癌精准诊疗再“升级”:首部《EG
作者:雨过天晴
肺癌治疗已进入“精准时代”,靶向药物让许多EGFR突变患者获得了更长
肺癌精准诊疗再“升级”:首部《EG
作者:雨过天晴
肺癌治疗已进入“精准时代”,靶向药物让许多EGFR突变患者获得了更长
 史美祺教授、李咏生教授:MET扩增精
整理者:雨过天晴审核人:鹰版为帮助MET基因异常等少见靶点NSCLC患者解决在治疗过程中
史美祺教授、李咏生教授:MET扩增精
整理者:雨过天晴审核人:鹰版为帮助MET基因异常等少见靶点NSCLC患者解决在治疗过程中
 10个月伏美和谷美耐药了,求助新方案
母亲 55岁 161 53kg 低分化腺癌晚期
查出来的时候脑转 骨转 肝转
24年11月底基因检
10个月伏美和谷美耐药了,求助新方案
母亲 55岁 161 53kg 低分化腺癌晚期
查出来的时候脑转 骨转 肝转
24年11月底基因检





 显身卡
显身卡