Targeting RAS and PI3K in lung cancer
Two crucial interlinked growth and survival signaling pathways have received enormous attention as targets for tumor therapy. One involves PI3K and downstream targets such as the protein kinase Akt (also known as protein kinase B). The other contains RAF kinases and their target kinase cascade, including mitogen-activated protein kinase kinase (MEK) and extracellular signal–related kinase (ERK). Both of these pathways are regulated by RAS proteins, which directly stimulate the activities of both PI3K and RAF
1. This bifurcated system (
Fig. 1) is constitutively activated in a high proportion of human tumors as a result of frequent mutations in several components of the pathways.
Figure 1: The Ras and PI3K signaling pathways.
Growth factors activate RAS through receptor tyrosine kinases, leading to stimulation of the RAF-MEK-ERK kinase cascade and also the PI3K pathway and other pathways involved in the control of cellular growth, proliferation and survival. Activation of the signaling network commonly occurs in human tumors through mutation of the components shown in blue or the deletion of those in yellow. Engelman
et al.
3use drugs (purple) targeting PI3K to successfully treat lung tumors induced by PI3K in a mouse transgenic model. They also use a combination of PI3K and MEK inhibitory drugs to block the growth of lung tumors in a RAS-driven mouse transgenic model.
Full size image (78 KB)
Small molecule inhibitors of PI3K have entered into early clinical trials in the past year or so
2, but it will be some time before clear indications of efficacy against human cancer emerge. In the meantime, large bodies of preclinical data have been acquired with these agents, including data from xenografts of human tumors in immunodeficient mice. Some of these studies have been encouraging, but until now their effects have not been tested in animal models that could be considered to approximate to the human disease.
The findings of Engelman
et al.
3 in this issue of
Nature Medicine fill in this gap. They show that a newly developed drug targeting PI3K reverses lung tumor formation in a transgenic mouse lung cancer model in which mutant PI3K is induced
3. Furthermore, they show that although this drug by itself is ineffective against Kras-induced mouse lung tumors, when combined with an inhibitor of the MEK pathway, it can cause regression of this most stubbornly resistant of tumor types
3.
Activating mutations in the
PIK3CA gene (encoding the type I PI3K
catalytic subunit) have been discovered in large numbers of human tumors
4, including 27% of breast and 19% of colon cancers (according to the Catalogue Of Somatic Mutations In Cancer database,
http://www.sanger.ac.uk/genetics/CGP/cosmic/). Such findings have led to a huge upturn in interest in PI3K as a drug target in cancer over the past 5 years. This interest has been further primed by findings that other molecules in the pathway are also frequently affected in cancers, including the
BRAF gene, the
PIK3CA gene and the
PTEN gene, encoding the lipid phosphatase that reverses the effects of PI3K. For example,
PTEN is deleted in about 10% of lung and colorectal cancers—although it is not clear that PTEN loss and PI3K mutational activation are equivalent in terms of disease outcome.
Engelman
et al.
3 used a transgenic approach to inducibly express an activated point mutant form of PIK3CA in the mouse lung. Although the lung is not the most obvious choice of target tissue for a PI3K cancer model, a low percentage of human lung tumors do show PIK3CA mutations, and they also have more frequent
PTEN deletions. The mice develop lung adenocarcinomas as quickly as 12 weeks after induction of transgene expression, allowing the testing of the pan-PI3K inhibitor NVP-BEZ235, currently in phase 1 and 2 trials
5. Established tumors in the mice regressed rapidly after treatment with the drug, an effect that could be separated from its ability to also inhibit the PI3K-related signaling hub mammalian target of rapamycin (mTOR).
Do these findings mean that human tumors carrying mutant PIK3CA or deleted
PTEN will respond favorably to NVP-BEZ235? Not necessarily. Mouse transgenic models of cancer are by nature simplistic approximations of the human disease. Tumors in these models are driven to develop rapidly by the strong activation of a single pathway upon which they tend to remain dependent—as also shown in this study by withdrawal of the transgene inducer
3. The extent to which most common human tumors remain dependent on the continued activity of initiating oncogenes (termed oncogene addiction) is still a matter of debate. However, the findings are certainly very encouraging and prove that the drug can potently reverse PI3K pathway activation within established tumors without generating limiting toxicity to the animal.
Engelman et al.3 also looked at the effect of NVP-BEZ235 on a mouse lung cancer model driven by the activated Kras oncogene. Genes in the RAS family are very frequently mutated in human cancer, including in 20–30% of lung cancers, and have evaded all attempts at therapeutic targeting so far. KRAS mutation is also a strong predictor of nonresponsiveness to epidermal growth factor receptor–targeted agents in lung and colon cancer. However, work from my own lab has suggested that RAS-mutant tumors might be sensitive to inhibitors of PI3K, one of its downstream interactors6, as mice in which PI3K was mutated so as to be unable to interact with RAS did not develop RAS-induced tumors. Engelman et al.3report a similar effect on Kras-driven lung tumor development in mice mutant for the regulatory subunits of PI3K, and their data from mouse xenograft models also point in the same direction7.
Despite this encouraging genetic data, Engelman
et al.
3 found that NVP-BEZ235 treatment alone did not cause regression of
Kras-induced lung tumors. Many reasons for these apparently conflicting results are possible. Among the most obvious is that the development of a new tumor (studied by the genetic experiments) and the maintenance of an existing tumor (affected by drug use) are not the same thing at all.
Engelman
et al.
3 carried on by combining the PI3K inhibitor with an inhibitor of MEK, a member of one of the other pathways activated by RAS via RAF. They used the inhibitor ARRY-142886, also known as AZD6244, an inhibitor of MEK1 and MEK2 in phase 1 clinical trials
8. The inhibitor alone had modest effects on the
Kras-induced lung tumors, but, together with the PI3K inhibitor, it caused substantial regressions.
The synergistic effect of inhibition of the PI3K and MEK pathways on the reversal of RAS-induced transformation has been noted many times in cultured cells
in vitroby both pharmacological and genetic approaches
1. Similar effects have occasionally been glimpsed in xenograft models of established human cancer cell lines. However, the importance of seeing dramatic tumor regression for the first time with combinations of clinically tested drugs in a transgenic mouse model of RAS-driven cancer maintenance should not be underestimated.
The work of Engelman
et al. shows that, although RAS has remained obdurately resistant to direct assault, its actions can be tamed by combination targeting of its multiple downstream pathways. These promising combination approaches to cancer therapy will ultimately receive decisive testing in the clinic. In the meantime, it is to be hoped that the issue of their generality will be addressed in several of the other high-quality mouse models of RAS-induced cancer, perhaps most importantly in pancreatic cancer
9, where KRAS mutation rates reach 70% in the human disease.
归纳一下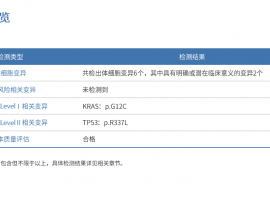 男77周岁 肺腺癌 2018年右上肺切除
父亲77岁 2018肺腺癌右上肺切除,今年6月份增强CT检查报告(右上肺术后,术区见金属缝
男77周岁 肺腺癌 2018年右上肺切除
父亲77岁 2018肺腺癌右上肺切除,今年6月份增强CT检查报告(右上肺术后,术区见金属缝
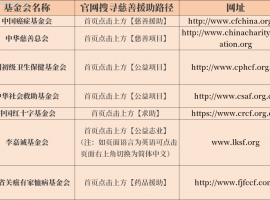 抗癌报销80%以上!学会这5个方法都能
作者:pear抗癌是一场与时间赛跑的持久战,对于患者家庭而言,除了直面疾病带来的挑战
抗癌报销80%以上!学会这5个方法都能
作者:pear抗癌是一场与时间赛跑的持久战,对于患者家庭而言,除了直面疾病带来的挑战
 23年脑膜转,目前脑部稳定,肺原发灶
2025.3至2025.5.22脑实质和脑膜稳定,肺部原发灶及胸腔积液控制不住。CEA持续升高。
23年脑膜转,目前脑部稳定,肺原发灶
2025.3至2025.5.22脑实质和脑膜稳定,肺部原发灶及胸腔积液控制不住。CEA持续升高。
 林根:小细胞肺癌放疗及免疫治疗常见
整理者:雨过天晴审核人:鹰版小细胞肺癌(SCLC)是一种恶性程度高、侵袭性强的肺癌亚
林根:小细胞肺癌放疗及免疫治疗常见
整理者:雨过天晴审核人:鹰版小细胞肺癌(SCLC)是一种恶性程度高、侵袭性强的肺癌亚
 求助:肺腺癌3年,脑转脑膜转骨转,E
肺腺癌3年,脑转脑膜转、骨转,E19+797S突变,免疫阴性,阿美29个月,期间放疗2次,后
求助:肺腺癌3年,脑转脑膜转骨转,E
肺腺癌3年,脑转脑膜转、骨转,E19+797S突变,免疫阴性,阿美29个月,期间放疗2次,后





 显身卡
显身卡





